

Drug discovery is the process by which new medications are discovered. Historically, drugs were discovered after screening by identifying the active ingredient from herbal remedies or crude extracts from plants or microorganisms suspected to have potential biological activity without the knowledge of their biological target. Only after the active principle has been identified was an effort made to identify the biological target. This approach to drug discovery is known as classical pharmacology.
It is understood that individual chemicals are required for the biological activity of a drug. This is based on the premise that drugs mediate their effect in the human body by specific interactions of the drug molecule with its target – biological macromolecules, such as proteins or nucleic acids in most cases. A drug target is the naturally existing cellular or molecular structure involved in the physiological/pathological pathway of interest that the drug-in-development is meant to act on.
Modern approach to drug discovery is based on the understanding of the aforementioned principle of drug action, employing the use of High Throughput Screening (HTS) of large chemical libraries of synthetic small molecules, natural products or extracts against isolated biological targets to identify a compound that is capable of eliciting the desirable therapeutic effect in a process known as reverse pharmacology. The method is the most frequently used approach today and has the advantage of requiring minimal compound design or prior knowledge. Although traditional HTS often results in multiple hit compounds, some of which are capable of being modified into a lead and later a novel therapeutic, the hit rate for HTS is often extremely low.
It is generally recognised that this approach to drug discovery and development (the use of HTS) is a time and resource consuming process. As noted by Anson et al (2009) despite advances in technology and understanding of biological systems, current approach to drug discovery is still a lengthy “expensive, difficult, and inefficient process” with low rate of new therapeutic discovery. Estimates of time and cost of currently bringing a new drug to market vary, but 7–12 years and $ 1.2 billion are often cited. In 2010, it was estimated that the cost of research and development of new molecular entities (NME) was US$1.8 billion. Thus, it is evident that pharmaceutical industry needs to find means of improving efficiency and effectiveness of drug discovery and development in order to sustain itself.
New approach
An emerging approach to drug discovery, involving the use of computing power to streamline drug discovery and development process, is rapidly gaining popularity and shows some promise in reducing time and cost in drug discovery process. Various terms are being applied to describe this approach, including Computer-Aided Drug Design (CADD), Computational Drug Design, Computer-Aided Molecular Design (CAMD), Computer-Aided Molecular Modelling (CAMM), Rational Drug Design, In Silico Drug Design, and Computer-Aided Rational Drug Design.
This approach leverages on chemical and biological information about ligands and/or biological targets to identify and optimise new drugs. This has been made possible by increase identification of molecular targets, elucidation of the 3D structures by X- ray crystallography and nuclear magnetic resonance (NMR), availability of commercial, private or public data bases (of biological targets and ligands) and availability of computer-aided drug design softwares.
CADD employs the use of in silico filters to identify hits (active drug candidates), eliminate compounds with undesirable properties (poor activity and/or poor Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET), selects the most promising candidates for further evaluation, and optimises these leads i.e. transform biologically active compounds into suitable drugs by improving their physicochemical, pharmaceutical, ADMET/PK (pharmacokinetic) properties.
A successful CADD campaign will allow identification of multiple lead compounds. Lead identification is often followed by several cycles of lead optimisation and subsequent lead identification, using CADD. Lead compounds are tested in vivo to identify drug candidates.
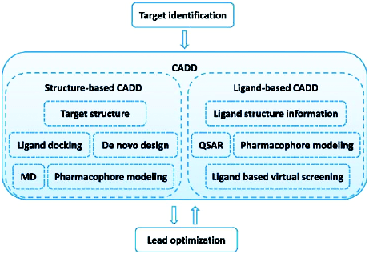
How it works
The process of drug discovery, using CADD approach begins with the identification of a therapeutic target against which a drug has to be developed. Depending on the availability of structural information, a structure-based approach or a ligand-based approach is used. Structure-based computer aided drug design depends on the information of the target protein structure obtained from X-ray crystallography, NMR or homology modelling to calculate interaction energies for all tested compounds. This approach involves “docking” a process of ligand binding to its receptor or target protein, to identify and optimise drug candidates by examining and modelling molecular interactions between ligands and target macromolecules.
Ligand-based computer-aided drug design approach, on the other hand, is used when 3D structural information of the target is not available and involves the analysis of compounds known to interact with a target of interest. This method relies on the Similar Property Principle, published by Maggiora and Shanmugasundaram (2011). It states that molecules that are structurally similar are likely to have similar properties. Structural properties considered in this approach include molecular weight, surface areas, ring content, atom types, electro-negativities, atom distribution, interatomic distances, bond distances, planar and non-planar systems, rotatable bonds, symmetry, functional group composition, aromaticity, solvation properties, and many others.
The overall goal is to characterise compounds in such a way that the physicochemical properties most needed for their desired interactions are retained, whereas unnecessary information not relevant to the interactions is discarded. It is considered an indirect approach to drug discovery in that it does not necessitate knowledge of the structure of the target of interest.
Advantages
According to Kapetanovic (2008) computational drug design expedites and facilitates the process of drug discovery from target identification, hit identification, hit to lead selection and lead optimisation. It increases the effectiveness and efficiency of drug discovery at a lower price, compared to the conventional drug discovery and decreases the use of animals in the process of lead identification and optimization.
Another benefit of in CADD is application in the screening of virtual compound libraries, also known as virtual High Throughput Screening (vHTS). This allows researchers to focus resources on testing compounds likely to have the activity of interest. In this way, a researcher can identify an equal number of hits while screening significantly less compounds, because compounds predicted to be inactive with high confidence may be skipped.
Avoiding a large population of inactive compounds saves money, time and resources, just as the pharma mantra goes, “fail fast, fail early”.
References
- Anson D, Ma J, He J-Q (May 2009). “Identifying Cardiotoxic Compounds”. Genetic Engineering & Biotechnology News. TechNote 29 (9) (Mary Ann Liebert). pp. 34–35. ISSN 1935-472X. OCLC 77706455. Archived from the original on 25 July 2009. Retrieved 25 July 2009
- Kapetanovic I.M. (2008) Computer-Aided Drug Discovery and Development (CADDD): In Silico-Chemico-Biological Approach. Chem Biol Interact. 2008 January 30; 171(2): 165–176. doi:10.1016/j.cbi.2006.12.006.
- Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL (Mar 2010). “How to improve R&D productivity: the pharmaceutical industry’s grand challenge”. Nature Reviews. Drug Discovery 9 (3): 203–14. doi:10.1038/nrd3078. PMID 20168317.
- Sliwoski G, Kothiwale S, Meiler J, and. Lowe E. W. (2014) Computational Methods in Drug Discovery. Pharmacological Reviews. 66:334–395.
- Wikipedia: Drug Discovery










{kind=link}